EU Clinical Trials Application Process

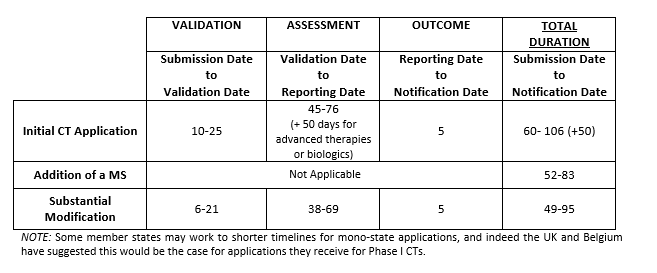
One of the major changes the EU Clinical Trial Regulation2 will introduce is a new clinical trial (CT) application procedure. Under the Regulation, Sponsors will be required to apply, via a new EU portal for authorisation to conduct an interventional clinical trial with medicines in Europe. The date the Regulation will apply is dependent upon the availability of the portal and the associated database which, according to the most recent EMA confirmation 1 , will be ready for use to support an effective date of “by October 2018 at the latest”. The Regulation introduces a single approach for the application and maintenance of the CT authorisation and this applies to trials whether they are multi-member state or mono-state. The Regulation defines procedural timelines (see Table 2) and is set to lead to harmonised document requirements for the EU and a reduction in the administrative burden of applications under the CT Directive. It introduces a procedure that not only combines the content of what is can be currently referred to as the “regulatory” and “ethics” applications, but also combines the scientific, technical and ethical review necessary to receive approval to conduct a CT in the EU. The application is submitted via the portal to all Concerned Member States (CMS) where the Sponsor intends to conduct the CT. At the time of application the Sponsor nominates a reporting member state (RMS). The RMS nomination may not always be granted and it is at Day 6 following submission that the RMS is confirmed. Important elements of the application process and subsequent updates:
- The application content (see Table 1) and the assessment are divided into two. Part I and Part II will be assessed in parallel unless the application only contains Part I.
- The Sponsor can submit only Part for assessment and may then submit within two years of the Reporting Date of Part I apply for an authorisation limited to aspects covered by Part II of the assessment report. In the MS for which the Sponsor does not submit within 2 years an application for authorisation to conduct the trial, the application in that MS will lapse.
- There is provision for cross-referencing to existing applications which will further reduce the current administrative burden of EU CT applications.
- The Regulation requires strict adherence to the maximum timelines allocated to each phase and has provisions for tacit withdrawal or tacit approval to ensure that delays from any party do not hold up the process.
- The timelines may result an increase or decrease to the overall timelines compared to current situation in some MSs, it will bring with it increased predictability to study start-up in the EU.
- A Sponsor may withdraw an application at any time until the Reporting Date but can only withdraw the full application rather than in chosen MSs.
- The addition of a member state in a modification to an approved application. The rMS will remain the same and the cMS will assess the application.
- Only substantial changes, known as modifications in the Regulation, require approval prior to implementation. The criteria for a substantial modification are described in the Regulation and are similar to the CT Directive. Substantial modification can be for Part I, Part II or both. Tracked changes version for modified documents may be required for the application.
- An application for a modification cannot be submitted if another is ongoing.
| Table 1. Summary of the Contents of the a new CT Application under the Regulation | |
| Part 1 Scientific and Medicinal Product Documentation | Part II National and Patient Level Documentation |
|
|
Diagram 1: EU Clinical Trials Application Process (Initial Applications)
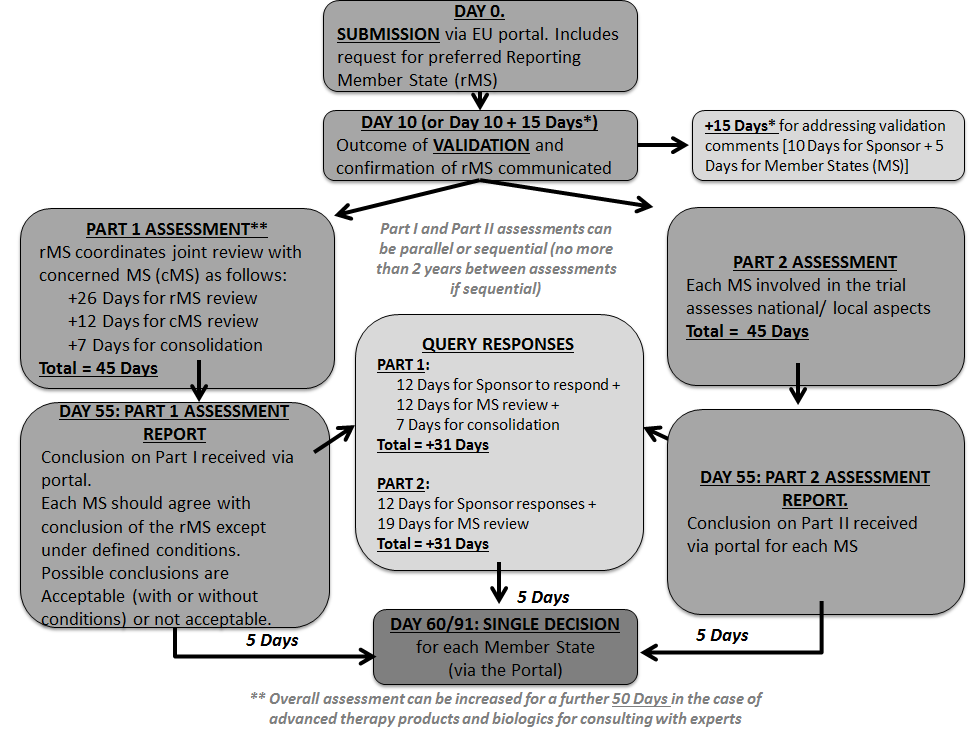
Table 2. EU Clinical Trials Application Process Timelines
- Introduction to EU Clinical Trial Regulation No. 536/2014
- Auxiliary Medicinal Products in EU Clinical Trials
- Clinical Labeling of Medicinal Products: EU Clinical Trial Regulation
Want to receive the next installment delivered straight to your inbox? Subscribe to ISPEAK.
Acknowledgements
Ted Bradley (Pfizer) – IP COP Task Team Contributor
Hans von Steiger (Pfizer) – Regulation Introduction Author
Magali Busqet (Sanofi) – IP COP Task Team Contributor
Massimo Eli (Merck) – IP COP Task Team Contributor
Chuck Gentile (Sanofi) – IP COP Task Team Contributor
Juliette Kirk (Pfizer) – IP COP Regulatory Consult
Kirsteen Magee (Mylan) – IP COP Task Team Contributor
Marianne Oth (Eli Lilly) – IP COP Task Team Contributor
Martin Waldherr (Roche) – IP COP Task Team Contributor



