iSpeak Blog
Are You Ready for an FDA Audit of Your Downstream Process?

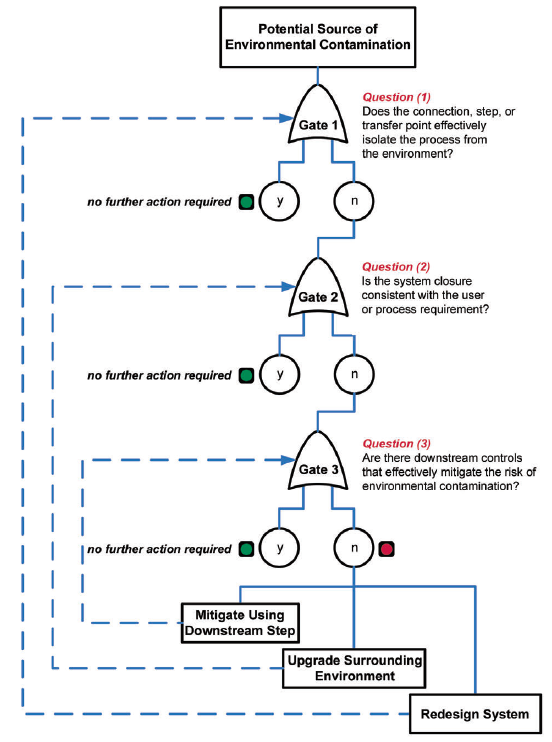
Figure 4.10: The Fault Tree Flow Diagram from ISPE Baseline® Guide for Biopharmaceutical Manufacturing Facilities
If an FDA inspector surprises you by asking questions about downstream microbial and endotoxin control, could you convince the inspector that you have adequate control over your downstream process to prevent a contamination? In recent audits, the FDA has taken a closer look at downstream microbial and endotoxin in process monitoring results, and are frequently requesting microbial and endotoxin results after validated hold-times. Not only is it important to have appropriate microbial and endotoxin testing data for equipment hold times and sanitization/sterilization validations, the FDA also wants confirmation that you have adequate proof of closure or room classification for each unit operation to minimize the risk of product contamination by potential adventitious agents.
Having a closed system that is completely isolated from the room environment has the lowest risk of contamination, but some steps require brief or prolonged exposure to the environment. For those steps, which are exposed to the environment, they must be performed in a room of adequate cleanliness or classification to protect the product or be further re-designed to decrease the risk of contamination (e.g. engineering controls). Section 4.0 from the ISPE Baseline® Guide for Biopharmaceutical Manufacturing Facilities (2013, 2nd Edition) describes sources of contamination and a methodology to perform a “closure analysis”. The “Closure Analysis” is a risk assessment which considers the closure scale (CS), bioburden control requirement or process requirement (PR), and calculates a risk rating (CS x PR = RR) for the unit operation. If the risk rating is calculated to be a 2 or 3, a moderate or high risk, then Figure 4.10 the Fault Tree Flow Diagram from ISPE Baseline® Guide for Biopharmaceutical Manufacturing Facilities is used to determine if the unit operation is appropriately closed in a suitable environment or needs to be redesigned or placed in a cleaner/ higher classification environment.
In summary, what is expected is documented evidence of process, equipment and environmental in-depth understanding about microbial controls and the rationale for the current design and use. First example, an open system aseptic operation such as inoculum preparation is required by the EU GMPs[i] is to be performed in Grade A environment, where as a closed aseptic operation with no environmental exposure such as fermentation can be performed in CNC environment. Second example, also required by the EU GMPs[ii], pre-viral reduction protein A affinity chromatography must be performed in a separate room from post-viral reduction ultrafiltration/diafiltration, if they are open systems. If closed systems, pre-viral reduction protein A affinity chromatography can be performed in the same room as the post-viral reduction ultrafiltration/diafiltration.
A well-documented closure analysis that proves your system is closed and/or being performed in a room of the appropriate classification, will bolster your in-process and hold time data, and provide additional evidence that your process is not at risk for microbial or endotoxin contamination. In addition to supporting audit inquiries, closure analysis is a powerful tool for evaluating process or facility changes Do not be surprised during your next audit, have a well-documented closure analysis ready!1 2


Through the ISPE Foundation Professional Development Grant program, Silas Tamufor attended the 2023 ISPE Annual Meeting & Expo in October 2023. Tamufor is a PhD student and ISPE Boston Chapter member who began serving as the ISPE Boston Educational Programs Committee Chair in December 2023. He, along with 87 other students and recent graduates, attended the conference thanks to the...

To meet the biopharmaceutical industry’s duty to manufacture safe and effective therapies for patients, a robust quality system is fundamental to success. A quality system should link to quality culture and prioritize focusing on quality, led by management, that fosters sustainable compliance and consistent production of high-quality drugs. Strong quality culture attributes include a proactive...

Stability sampling and testing are key to ensuring that products maintain safety, identity, strength, purity, and quality throughout their claimed shelf life. It is also a regulatory requirement per ICH Q5. However, storing product samples in different environmental conditions, testing those samples for three to five years (or more) after initial manufacture, and properly analyzing and...