Selection Criteria for Pharmaceutical Containment Equipment

Pharmaceutical manufacturing facilities produce a variety of products, including highly potent products that require safety measures to prevent adverse health effects on patients and operators. To ensure safety, these facilities use containment equipment to minimize the risk of contamination. This article presents criteria for selecting containment equipment, considering both cross-contamination and industrial hygiene risks.
Pharmaceutical manufacturing facilities produce a variety of products, including highly potent products that require safety measures to prevent adverse health effects on patients and operators. To ensure safety, these facilities use containment equipment to minimize the risk of contamination. This article presents criteria for selecting containment equipment, considering both cross-contamination and industrial hygiene risks.
Planning safety in pharmaceutical manufacturing facilities requires a proper evaluation of the risk involved, such as the risk of cross-contamination between the target product and other drugs and the risk of industrial hygiene to operators. Based on this proper evaluation, the appropriate containment equipment should be selected for use in manufacturing facilities.
Selection Criteria Overview
The selection of containment equipment is often based on the degree of airborne exposure because inhalation or airborne transfer are the main routes of exposure for operators. This selection method has proven effective in reducing industrial hygiene risks to operators.
Containment equipment is also effective in preventing cross-contamination risk in pharmaceutical manufacturing. It is important to consider expo-sure through mechanical transfer, in addition to airborne transfer, when evaluating containment equipment. However, there has not been a system proposed for selecting containment equipment based on the degree of exposure through mechanical transfer.
Selection criteria are based on the evaluation of exposure routes through both airborne and mechanical transfer and are driven by advancements in engineering for highly active pharmaceuticals and exposure measurement. This criterion could also be applied to maintenance design of the accessories for pharmaceutical manufacturing equipment or containment equipment, such as changing the filters of dust collector or fluid bed dryers.
The containment equipment selected during the initial design phase may need to be reselected as a result of more detailed risk assessments conducted later in the design process, such as failure modes and effect analysis (FMEA), hazard and operability studies (HAZOP), or other similar methods. In particular, the source of both cross-contamination and industrial hygiene issues, which are often due to the changeover for each campaign manufacture, needs to be evaluated in terms of standard operating procedures (SOPs) and equipment design.
The keystone of this article is organized in the following section, which introduces the risk assessment method that forms the basis of this article, and in the section on the method for selecting containment equipment, which delves into the specific steps for selecting containment equipment.
Risk Assessment Method
In developing a method for selecting appropriate containment equipment considering both cross-contamination and industrial hygiene risks, it is important to clearly identify each risk scenario and the acceptable level of risk.
Risk Assessment for Cross-Contamination
Risk is a function of severity and probability:
Risk = ƒ (Severity, Probability)
In applying this function, the risk of cross-contamination can be defined as the combination of the amount of drug A entering into drug B (amount of exposure) and the potential toxicity (hazard) of drug A that causes health problems in patients administered with drug B. In other words, cross-contamination risk is a function of both hazard and exposure:1
Cross-contamination Risk = ƒ (Hazard, Exposure)
Note that although hazard is defined as “the potential source of harm (ISO/IEC Guide 51:2014)” in the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use’s (ICH’s) Q9(R1),2 in this article, we define it as the toxicological potential of a drug to cause adverse health effects (toxicity).
Cross-contamination risk assessment in multiproduct facilities, in accordance with the quality risk management process of ICH Q9, identifies whether the risk of cross-contamination in each manufacturing process is acceptable or not. This is accomplished by a) identifying exposure routes of cross-contamination and analyzing the amount of exposure caused by that route and b) using the potential toxicity of the drug (cross-contamination limit) as an index.
According to ICH Q9(R1) 4.3 Risk Assessment: “In some risk management tools, the ability to detect the harm (detectability) also factors in the estimation of risk.”2 But detectability is often clarified during the detailed specification phase of containment equipment. Therefore, it was not considered in the stage of selecting the basic functionality of the containment device.
Identification of the cross-contamination exposure route
If cross-contamination of products occurs during the manufacturing process, patients who are administered that product will be exposed to the risk of adverse health effects caused by the other cross-contaminated drug. Identifying the cross-contamination route (risk factor) is identifying “a potential source of harm,” as mentioned in ICH Q9(R1).2
There are four routes that cause cross-contamination risks—mix-up, retention, mechanical transfer, and airborne transfer—and the respective risks are identified1 ,3 in the following sections.
Mix-up
Mix-up is the use of the wrong active pharmaceutical ingredients (APIs), intermediates, or products and is mainly caused by human error. GMPs and related guidelines have proposed various requirements to prevent mix-up. To reduce the risk of cross-contamination through this exposure route, implementing monitoring systems and measures to prevent human error (e.g., appropriate SOPs and labeling, double-checking) is effective. Containment equipment does not contribute to this risk reduction.
Retention
Retention is defined as the carryover of material on product contact surfaces from one product to another in the same equipment used in a campaign manufacturing. The factors affecting retention relate to the effectiveness of the cleaning procedure.1 That is, the cross-contamination risk due to retention has been accepted through cleaning validation. However, as specified in the 2015 revision of the PIC/S GMP Guide Annex 15 (Process Validation),4 the acceptable residue limits need to be established based on toxicological health-based exposure limits. This differs from industry-proposed limits, such as 10 ppm (parts per million), 1/1000, and the visual detection limit that have been adopted by many pharmaceutical companies.5 If the equipment is designed so that powder loading into the equipment, discharge of intermediate products from the equipment, or changeover cleaning (clean-in-place [CIP] compliance) can be performed under completely closed conditions, then the risk of cross-contamination due to retention is acceptable through cleaning validation. However, any type of equipment, especially for solid dosage forms, cannot maintain a completely closed state (containment of exposure below acceptable limits) for such processes. Therefore, it is important to consider the exposure risk from other routes such as mechanical and airborne transfer.
In developing a method for selecting appropriate containment equipment considering both cross-contamination and industrial hygiene risks, it is important to clearly identify each risk scenario and the acceptable level of risk.
Mechanical transfer
Mechanical transfer is an event in which a drug substance is transferred from a product contact surface to a nonproduct contact surface (e.g., the exterior surface of manufacturing equipment, facility walls and floors, operator’s garment) and later is mixed with other products. There is no need to consider cross-contamination due to mechanical transfer if the products are not transferred to a nonproduct contact surface.
However, many mechanical transfers occur in the pharmaceutical manufacturing process and all routes by which material can be transferred from contaminated nonproduct surfaces into the product must be considered. This includes product contact surfaces contaminated by contact with contaminated surfaces, inadvertent or transient contact with other contaminated non-designated product contact areas, and direct contact of the product with such surfaces as operator apparel and gloves.1
One example includes any solid dosage manufacturing equipment that handles powders, such as tableting machines. If the equipment cannot be cleaned to below acceptance level without disassembly, the disassembly of wet parts after wet in place, cleaning, and reassembly of that equipment presents a cross-contamination risk, due to the assumption that the drug substance would adhere to the operator’s garment.
If tableting machines and other formulation equipment could be cleaned in a completely closed state without requiring disassembly, like in CIP systems, then the risk of the cross-contamination caused by mechanical transfer would be acceptable. Physical containment of the product contact part, which is the source of contamination, is also effective in reducing the cross-contamination risk caused by mechanical transfer. The degree of cross-contamination risk varies depending on the robustness of the containment equipment.
Airborne transfer
Airborne transfer is an event in which the drug substance on the product contact surface becomes airborne and disperses in the form of aerosol or dust and carries over into another product. Physically segregating the product contact surface and containment by airflow is effective in reducing the cross-contamination risk by airborne transfer. The degree of cross-contamination risk varies depending on the selection of the containment equipment.
In other words, containment equipment cannot contribute to the reduction of cross-contamination through mix-up and retention, but it can be effective in reducing the risk of cross-contamination via mechanical transfer and airborne transfer.
Analysis of the cross-contamination limits
Cross-contamination shall be analyzed to determine whether the risk of cross-contamination of the manufacturing process is at an acceptable level by using the potential toxicity of a drug substance (cross-contamination limit) as an index. It is necessary to scientifically establish the cross-contamination limit based on toxicology or pharmacology.
A representative index of the health-based exposure limit is the permitted daily exposure (PDE)/acceptable daily exposure (ADE). ADE is defined as “a dose that is unlikely to cause an adverse effect if an individual is exposed, by any route, at or below this dose every day for a lifetime.”1 The PDE is established by identifying a threshold without adverse health effects based on validated preclinical (animal) and clinical (human) data and incorporating appropriate safety factors.
Note that the terms used in Europe and the United States to express ADE is different as follows, but the definition is the same toxicologically. In Europe, PDE is used by the European Medicines Agency, ICH, and the PIC/S. In the US, ADE is used by the FDA and ISPE.
Risk evaluation of cross-contamination exposure routes
In pharmaceutical manufacturing, cross-contamination risk of each exposure route should be evaluated using the following method. This will ensure that the APIs of a product manufactured in a shared equipment for multiproduct facilities do not enter into a daily dose of another product above its PDE level:
- In many cases, risk of cross-contamination due to mix-up can be reduced by detection through a specification test.
- For the risk evaluation of cross-contamination due to retention, cleaning validation is effective. The critical limits of cleaning validation, however, should be toxicological-based limits.
- Risk of cross-contamination due to mechanical and airborne transfer has been considered acceptable if the nonproduct contact surfaces (operation rooms, exterior surfaces of equipment) are visually cleaned. However, in manufacturing processes where highly active drugs are handled, the risk evaluation of cross-contamination using conventional visual inspection cannot be regarded as a risk evaluation based on scientific knowledge.
Although these cross-contamination risks were considered detectable through the specification tests, they cannot be detected by the verification of a limited number of samples. This is because contamination of another drug via mechanical transfer and airborne transfer usually occurs locally and does not uniformly contaminate the whole batch.
It is necessary to evaluate the cross-contamination and industrial hygiene risk through mechanical transfer and airborne transfer by:
- Identifying the route of carryover into another product
- Identifying the amount of carryover caused by that route
- Evaluating the acceptability of that amount of contamination using an acceptable limit as an index
It is reasonable to consider the exposure route that represents an exposure scenario based on actual cases. Thus, we should focus on airborne exposure from the boundary surface of containment equipment and exposure to residues on surfaces that operators could contact. The acceptable level is applied to the risk assessment of cross-contamination due to mechanical or airborne transfer previously described. Cross-contamination is often caused by operators contacting surface residues; therefore, adopting wipe limits (surface exposure limits), an industrial hygiene index that shows how operators contact with the contaminated surface, is considered reasonable.
However, the airborne exposure measurement at some specified points is not always lower than that of the exposure measurement for operators. This means that it is not enough to assess the risk of cross-contamination by merely evaluating the operators’ exposure. It is reasonable that the amount of the airborne exposure on the boundary of containment equipment can be considered the sum of the airborne exposures that could cause cross-contamination.
Risk Assessment for Industrial Hygiene
As one aspect of “industrial hygiene” as defined by OSHA,6 this article introduces the concept that to ensure operators’ safety, the amount of residue on surfaces with which they may contact should remain below exposure limits. Operators are exposed to drug substances through a variety of routes during a manufacturing process, such as nasal and oral inhalation, dermal absorption, and mucosal absorption.
The definition of risk stated previously is also applicable to industrial hygiene risk. The industrial hygiene risk in this article can be defined as the combination of the probability of occurrence of harm due to the drug substance which will come into contact with an operator and the health disorder raised by the drug substance extracted.
The same method used for cross-contamination risk evaluation can be applied to the industrial hygiene risk evaluation. So, the evaluation can be carried out as follows: a) identify the route of operator exposure to drug substances, b) analyze the amount of exposure through that exposure route, and c) determine the acceptability of industrial hygiene risk of the process using the potential toxicity of the drug as an index.
Identification of industrial hygiene exposure
The exposure of drug substances to the operators is limited to mechanical and airborne transfer. As mentioned previously, the use of containment equipment is effective in reducing the cross-contamination risk caused by both mechanical and airborne transfer. The selection of containment equipment also reduces industrial hygiene risks.
The use of personal protective equipment (PPE) is effective in reducing exposure to the operator, but it is considered a last resort or secondary protection.1 It protects only operators, and it does not mean the environment where the operators’ activity takes place has been controlled. In this arti-cle, the effectiveness of PPE is not considered in the selection of containment equipment.
Analysis and evaluation of exposure limits for industrial hygiene
Permissible exposure limits for industrial hygiene should be scientifically established based on toxicology or pharmacology. The limits are set for each route of exposure, but in principle, operators should not intake more than PDE/ADE of the drug substance from any route of exposure.
Permissible exposure limits for airborne transfer
Occupational exposure limits (OELs) set an acceptable airborne concentration at or below which no adverse health effects to operators are expected after a daily work shift of eight hours (one working day) where the target substance exists. It is calculated based on PDE, unless there is an excessive workload. It is often calculated with an assumption of OEL = PDE/10, according to the air intake volume (approximately 10 m3) of an operator during the eight-hour working shift. If the bioavailability by inhalation has been verified, it should be considered.
Permissible exposure limits for mechanical transfer
The residues of drug substances on the surface where operators may touch should not exceed the wipe limits. Wipe limit is defined as PDE/100 cm2 .3 This is because 100 cm2 is the mean value of the total number of palms area, or the number of human hands, that could touch the surface. The use of these limits was proposed because operators’ palms are most likely to contact the substances handled by their hands.
In the establishment of these indices, there are arguments to strictly evaluate the bioavailability for dermal absorption, the number of times the operator touches the surface, and secondary (indirect) exposure. However, using the index method is beneficial to conveniently evaluate the residues on exterior surfaces of equipment and surfaces of facilities from the viewpoint of the operators’ safety.
Equipment Selection Method
Selection Criteria Control Exposure Risk
As mentioned previously, the use of containment equipment is effective for reducing cross-contamination risk caused by mechanical and airborne transfer, but not caused by mix-up and retention. Both cross-contamination and industrial hygiene risks are caused by the transfer of drug substances from product contact surfaces to nonproduct contact surfaces through mechanical and airborne transfer. These risks depend on the following factors:
- Physical/chemical properties of the subject drug substance (e.g., state, volume)
- Characteristics of the process operation (e.g., operations that apply energy to the subject drug substance resulting in airborne transfer, such as milling, or operations that cover the exposed surface, such as coating)
- Performance of containment equipment or containment performance of manufacturing equipment
In pharmaceutical manufacturing, it is impractical to change the formulation, the physical and chemical properties of the drug substance, or the manufacturing process unless in the development stage of new drugs. Therefore, selecting appropriate containment equipment is practically effective to control the industrial hygiene and cross-contamination risks.
Equipment Selection Policy
Basic policy for the selection of containment equipment should be established with the following procedure:
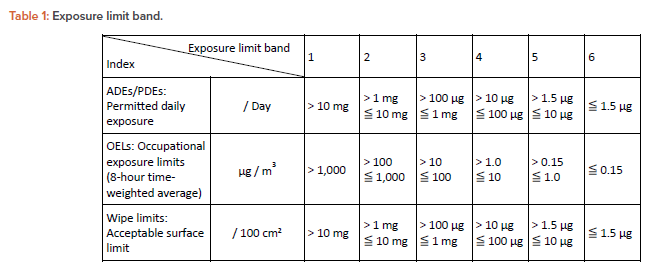
- Set permissible exposure limits: Establish the exposure limit band for industrial hygiene/cross-contamination risks based on the hazardous properties of the subject drug substances.
- Set exposure level: Identify the predicted exposure (PE) band. The details are described in the following paragraphs for each process operation based on the physical/chemical properties of the drug substances and the characteristics of process operations.
- Select engineering control (EC) band: Select an EC band to reduce the exposure of each identified process operation to a level below a permissible exposure level and select an appropriate containment equipment.
Each procedure is detailed in the next section.
Note 1:
The criteria for setting exposure limit band are as follows:
- OEL: 10 g/m3 is the occupational exposure limit level that the physical segregation should
consider - PDE: 1.5 g/day is the threshold of toxicological concern (TTC) value for mutagenic
substance - Wipe limits: 100 g/100 cm2 is the minimum visible detection limit
- OEL: 100–1,000 g/m3 is the range of visual detection limit for dust
Note 1:
When handling mutagens and anticancer drugs with genotoxicity, the most robust containment equipment for manufacturing equipment is used. Moreover, the ICH M7 guideline explains that the default value of PDE for the “compounds having direct e ect on DNA, those are, mutagens and anticancer drugs with genotoxicity,” is 1.5 g/day. This is based on the TTC except for the afl atoxin-like compounds, N-nitroso compounds, and alkyl azoxy compounds, which are classified into a group of highly mutagenic carcinogens “Cohort of Concern.”7 Therefore, a value of < 1.5 g/day is set for the PDE of the most active category in the exposure limit band.
Equipment Selection Procedures
Setting permissible exposure limits
Classifying the subject drug substance according to its exposure limits and sharing this with the related personnel ensures they understand that the materials they handle are hazardous property. Table 1 summarizes classified exposure limits, which we call the exposure limit band.
Setting the exposure level
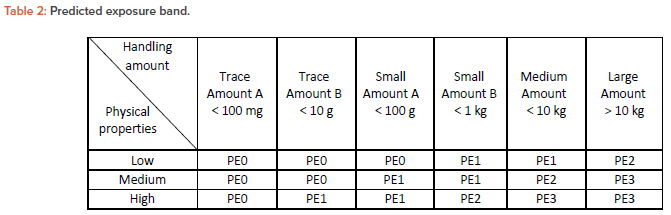
The subject material changes its physical form as the process operation proceeds and is exposed to a nonproduct contact surface from a product contact surface. The potential degree of exposure is predicted from the physical condition and the amount being handled, and a PE is determined based on the criteria described in Table 2.
Predicted exposure band
A PE band is used to classify the degree of exposure of a subject drug substance to nonproduct contact surface based on its physical form and daily handling amount. This index shares the same basic concept with the exposure predictor (EP) band proposed in Control of Substances Hazardous to Health Regulations (COSHH) Essentials.
In this article, however, airborne and the mechanical transfer are included when considering the degree of exposure of the subject substance to the nonproduct contact parts. In this respect, the PE band in this article is a different concept from EP in COSHH Essentials, and therefore the term PE is used.
Physical properties of the drug substance
Intermediate products change their physical properties as the process proceeds. The changes occur before and after each process. Therefore, physical properties of the substances handled in each process are determined at the inlet of the intermediate product to the process, during process, and at the outlet of the intermediate product. When applying Table 2, the physical properties are classified in the following categories, which define the viewpoint of the degree of exposure in the representative physical form of the drug substance.
These are the physical properties of drug substances in the filling process:
- Low: Liquid/suspension, soft capsules after dedusting, and coated tablets
- Medium: Uncoated tablets, wet powders, wet pellets, soft capsules before dedusting, hard capsules that are difficult to dedust, and coated granules
- High: Micropowders, dry powders, powders, fine granules, granulated products, and freeze-dried products
The degree of exposure during filling operation should be evaluated for each risk scenario, such as exposure in the event of bottles accidentally toppling. This is because the degree of exposure during the filling of the drug substance into containers depends on unexpected (but possible) accidental events in the process operation. It should be noted that what is important is evaluating the amount of daily exposure and not discussing the frequency of occurrence.
Handling amount
The handling amount in Table 2 indicates the daily amount of subject drug substance handled in that containment equipment because permissible exposure limits are based on a unit of one day. If the API is diluted with excipients, such as intermediates for solid dosage, the apparent exposure limit band can be used. In this case, the exposure limit band can be relaxed in accordance with a dilution rate as long as the actual amount of the API in the airborne dust correlates to the dilution ratio.
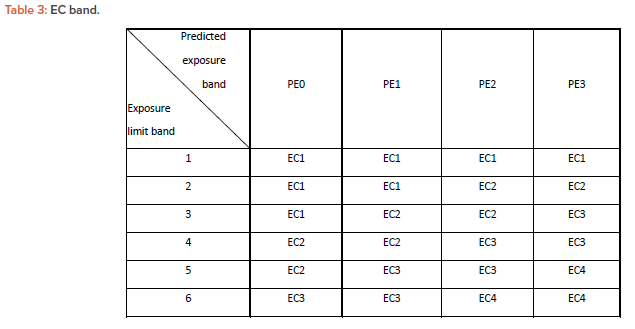
EC Band
Subject drugs should be handled at exposures below the permissible limit for each process operation. So, appropriate containment functions are required according to the physical properties and amounts handled and the exposure limits in the process. The containment function required is achieved by having equipment and facilities that control or limit transfer to nonproduct contact surfaces due to process operations.
The typical containment equipment used in the formulation process in the pharmaceutical industry can be classified into the following four EC band categories based on its containment performance during process operation and decontamination of the drug substance residues.
- EC1: General ventilation (HVAC) facility.
- EC2: Containment by air flow and local dedusting device. For example, local dust collection (external device), local dust collection (enclosed type, push-pull system), local dust collection (enclosed type, draft chamber type), closed restricted access barrier system.
- EC3: Physical isolation device, containment equipment using airflow that allows decontamination of the exposed area only by manual hand operation. For example, enclosed dust collection booth that allows in/out transfer of items and inside decontamination by hands, safety cabinet with unidirectional flow, negative pressurized isolator box with pass-through hatch or positive pressurized robust isolator, split valve, flexible (single use) isolator box.
- EC4: Physical isolation device that allows in/out transfer of items and inside decontamination under closed condition: For example, negative pressurized glovebox or negative pressurized isolator with rapid transfer port (RTP).
The containment equipment cannot be considered robust if the drug substance or operator is exposed to the outside of the chamber by air when de-contaminating the inside of the chamber or the drug substance is exposed to the outside of the chamber by mechanical transfer. This is true even if the equipment can control the containment by airflow.
For example, the equipment shown in Figure 1, which includes a structure to enclose the source of dust generation and controls exposure of open-ings for manual operation by airflow, may generate turbulence and vortex airflow in the chamber. The drug substance may be airborne to the area where the operator’s hands cannot reach. So, the operator may put their upper body inside the chamber to perform the decontamination work, and therefore, the EC band should be set to EC2.
On the other hand, in the airflow containment equipment shown in Figure 2, the airborne dust and adhesion of residues are limited because there is a laminar flow inside the chamber. The operator can decontaminate the chamber to a level below an acceptable residue limit by simply reaching out their hands into the chamber. When considering a series of process operations including decontamination, the containment performance is more robust than the above-mentioned equipment, and the EC band is set to EC3.
Fig 2
Selecting containment equipment
The exposure in each process operation is predicted and allocated into an appropriate EC band to control the exposure of a subject drug substance to the level below its acceptable limit. Table 3 is an EC band showing the containment categories determined based on the exposure levels and exposure limits predicted from the past exposure measurements. The EC band can be selected from the PE band for each process and exposure limit band. (A detailed explanation on how to create Table 3 is provided in the Appendix.)
Additional requirements for visual detection limits
Cross-contamination and industrial hygiene risks can be reduced if the degree of exposure can be visually detected. Visual detection limits are highly dependent on the characteristics of drug substance, the manufacturing process, and the internal surface specifications in the operation room.
The following requirements should be considered in the process validation and verification. In the case where the drug or intermediate is classified as 3–4 in the exposure limit band and the PDE/100 cm2 or under cannot be confirmed visually, identify the area where there is a potential for exposure in the actual manufacturing process or in the process simulation test using the surrogate powders. If necessary (e.g., if the exposure level exceeds 1/10 of the acceptable limit), the residuals should be verified on a periodic basis.
In the case where the drug or intermediate is classified as 4–6 in the exposure limit band, identify the area where there is a potential for exposure in the actual manufacturing process or in the process simulation test using the surrogate powders. If necessary (e.g., if the exposure level exceeds 1/10 of the acceptable limit), secondary exposure prevention measures should be considered, and the residues should be verified periodically. If the decontamination operation is performed manually, the residues inside the containment equipment (primary barrier) should be verified for every lot (or every campaign).
Conclusion
This article introduces a methodology for selecting containment equipment in the early stages of design for pharmaceutical facilities. Selecting contain-ment equipment does not mean that the cross-contamination or industrial hygiene risks of pharmaceutical manufacturing facilities are now acceptable. There are some pharmaceutical processes where adequate containment equipment cannot be applied.
In addition, the containment equipment is only a primary barrier, and a secondary barrier should be considered in case of primary barrier failures. Such comprehensive risk assessment should be defined and implemented as cross-contamination or industrial hygiene risk management based on ICH Q9 or a contamination control strategy.
In the risk assessment of containment equipment, it is also necessary to consider the following issues:
- Risk assessment of containment equipment specifications
- Risk assessment of secondary barriers, such as specification of facilities such as heating, ventilation, and air-conditioning (HVAC) systems and specification of the operation room (capabilities of containment, washing, decontamination)
- Risk assessment in case of containment equipment failure where PPE or emergency mist/shower would be needed
- Risk assessment in cases where containment equipment cannot be applied and technical/organized measures of a robust secondary barrier system would be needed
It is equally important to define their risk assessment methods, which may be introduced in a separate article if the opportunity arises.
This article includes major technical suggestions that can be selected and applied when establishing a quality management system (QMS) based on ICH Q9(R1). However, when incorporating these suggestions into an ICH Q9–based QMS, appropriate selection and adjustments may be required to reflect the actual situation of each manufacturing site. 8 , 9 , 10 , 11 , 12 , 13 , 14 , 15 , 16 , 17 , 18 , 19
Adjustment for the dust emission property of the process operation:
When assuming the degree of exposure for processes that amplify the energy for powders to become airborne, such as milling, it is not possible to select the appropriate containment equipment if the exposure band is set based on the physical property of the drug substance in Table 2. For such processes, therefore, the EC category selected based on the Table 3 EC band should be raised to one higher category.
Appendix: Method for Creating Table 3
Table 3.1
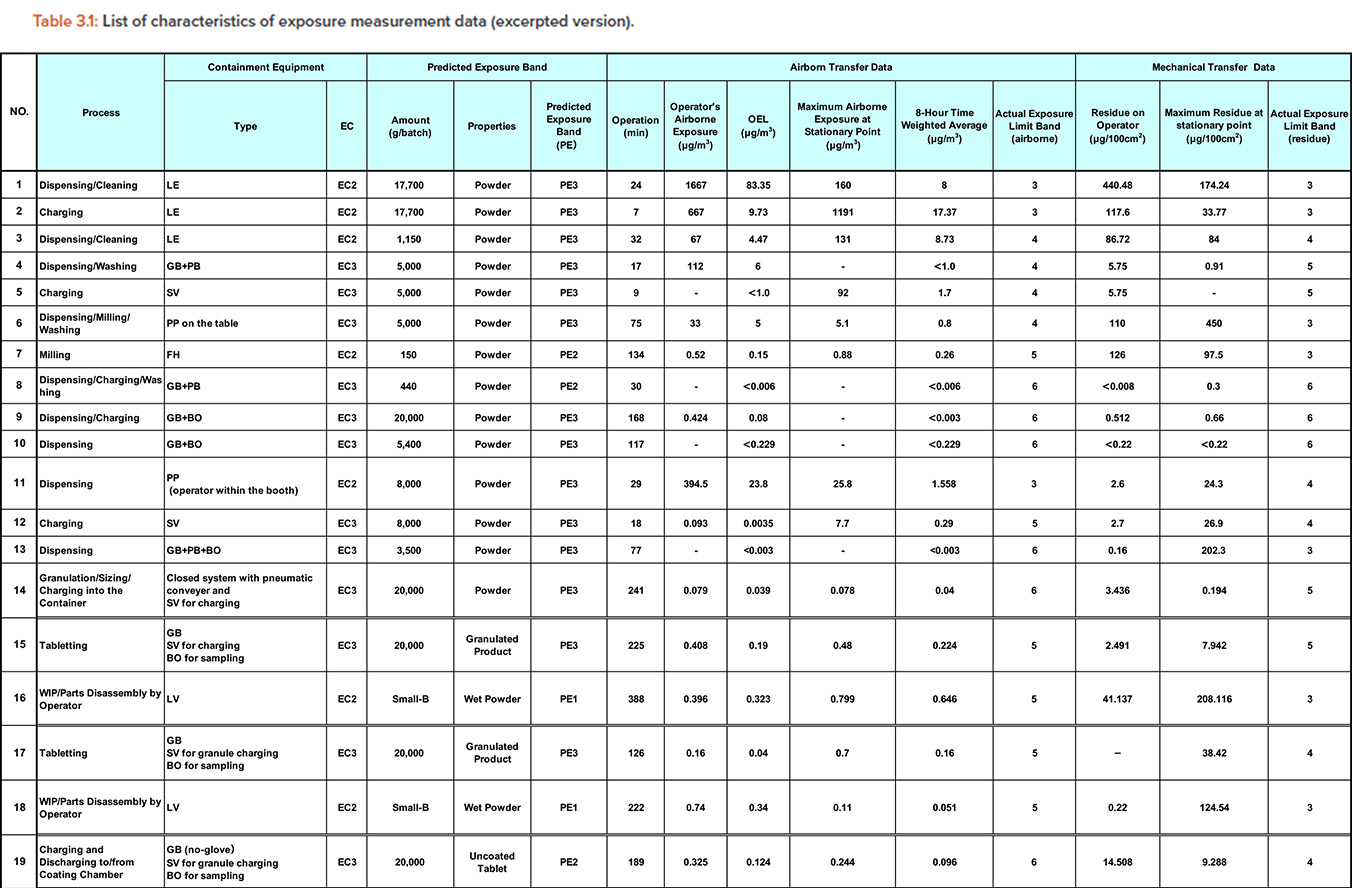
First, we prepared Table 3.1 from approximately 100 data points of our actual exposure measurements and actual exposure measurements shown in the published literature.
Data
The data in Table 3.1 included:
- Airborne exposure measurement data at operator’s mouth, which is shown as “operator’s airborne exposure.”
- Airborne exposure measurement data at fixed points around the containment equipment, which is shown as “maximum airborne exposure at stationary point.”
- Mechanical transfer measurement data remaining on operator’s chest in the form of residues, which is shown as “residue on operator.”
- Mechanical transfer measurement data remaining at fixed points around the containment equipment in the form of residues, which is shown as “maxi-mum residue at stationary point.”
Columns
- The containment equipment column shows the EC band, which represents the required containment performance for each process.
- The PE band column shows:
- The handling amount when measuring exposure
- The state of material to be handled
- The PE band selected from Table 2
- The airborne transfer data column shows:
- The operation time, operator’s airborne exposure, and OEL given by OEL = (operation time) × (operator’s airborne exposure) / (480 minutes)
- The maximum airborne exposure at stationary point and 8-hour time weight average given by: 8-hour time weight average = (operation time) × (maximum airborne exposure at stationary point) / (480 minutes)
- The actual exposure limit band (airborne), which is obtained by applying the largest value of OEL and 8-hour time weight average to Table 1
- The mechanical transfer data column shows:
- The residue on the operator, which is measured on the operator’s chest and maximum residue at stationary point
- The actual exposure limit band, which is obtained by applying the largest value of residue on operator and maximum residue at stationary point to Table 1
Abbreviations: BO: bag-out; DB: down-flow booth; EC: engineering control; FH: fume hood (draft chamber); GB: glovebox and isolator; HF: half-suit; LE: local exhaust; LV: local ventilation; OEL: occupational exposure limit; PB: pass box; PE: predicted exposure; PP: push-pull booth; SC: safety cabinet; SV: split valve; WIP: wet-in-place.
Tables 3.2 and 3.3
Second, we prepared Table 3.2 and Table 3.3.
Table 3.2 is created for airborne exposure with exposure limit band and EC band as the vertical axis and PE band as the horizontal axis. The number in each cell is the number of exposure measurement cases listed in Table 3.1, which is counted as one case per row.
Similarly, Table 3.3 is created for exposure by mechanical transfer with exposure limit band and EC band as the vertical axis and PE band as the hori-zontal axis, and the number in each cell is the number of exposure measurement cases listed in Table 3.1, which is counted as one case per row.
For example, the dispensing/cleaning process in the first row of Table 3.1 shows a dispensing/cleaning operation that assumes an exposure equivalent to PE3 for a containment equipment equivalent to EC2. From the airborne exposure at that time, we obtained the measurement result that we can handle substances up to exposure limit band 3. This example corresponds to the column of exposure limit band 3, EC = 2, and PE = 3 in Table 3.2.
Similarly, from the exposure by mechanical transfer, we obtained the measurement results that the materials up to exposure limit band 3 can be handled. In other words, this case corresponds to the column of exposure limit band = 3, EC = 2, and PE = 3 in Table 3.3.
In this example, the exposure limit band of the material handled in the dispensing/cleaning process is the same for airborne exposure and mechanical transfer. However, in other measurement data, the exposure limit band may differ depending on the exposure route. This indicates that the selection of containment equipment based solely on conventional airborne exposure is insufficient.
To indicate which column of Table 3.2 or 3.3 corresponds to which number of the data listed in Table 3.1, the data numbers are listed as circled numbers. In the same way, about 100 measurement cases were applied to Table 3.2/3.3.
For each exposure limit band (1-6) and PE band (PE0-PE3), selecting the EC band (EC1-4) corresponding to the shaded cell or the cell below it would result in the risk of cross-contamination and industrial hygiene to be accepted.
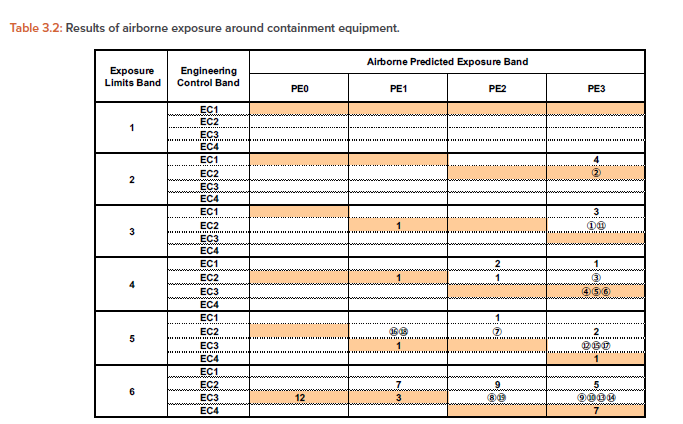
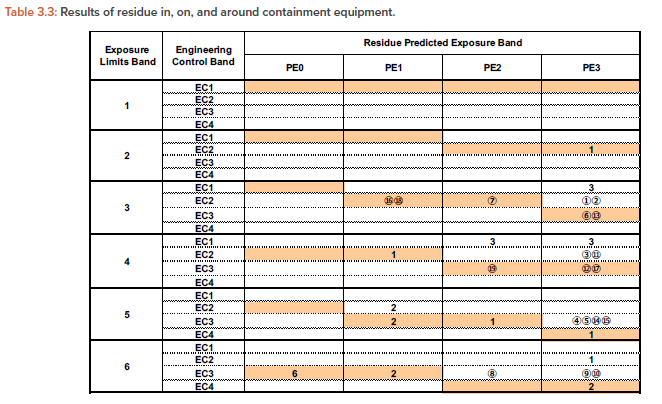
Table 3
Table 3 was established by summarizing the exposure limit band/EC/PE combinations corresponding to the masked cells. The data were not sufficient to fill all cells for the matrix, so Table 3 was created on the premise that cells with no exposure data available can be filled by interpolating from the existing exposure data. The exposure data in Table 3.2 and Table 3.3 indicate that containment to a level below acceptable exposure may be achieved even with containment equipment which has less performance than defined in Table 3. This means that Table 3 may be rather conservative. This table will be developed to be more precise and rational as future exposure data will be accumulated.
- 1 a b c d e f International Society for Pharmaceutical Engineering. ISPE Baseline® Guide: Risk-Based Manufacture of Pharmaceutical Products (Risk-MaPP), 2nd ed. North Bethesda, MD: International Society for Pharmaceutical Engineering, 2017.
- 2 a b c International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use. “ICH Harmonised Guideline Q9(R1): Quality Risk Management.” Final version adopted on 18 January 2023. https://database.ich.org/sites/default/files/ICH_Q9%28R1%29_Guideline_Step4_2023_0126_0.pdf
- 3 a b Mulhausen, J., and J. Damiano. A Strategy for Assessing and Managing Occupational Exposures. Fairfax, VA: The American Industrial Hygiene Association, Exposure Assessment Strategies Committee, AIHA Press, 1998.
- 4European Commission. “EudraLex, Volume 4: EU Guidelines for Good Manufacturing Practices for Medicinal Products for Human and Veterinary Use. Annex 15: Qualification and Validation.” Published 2015. https://ec.europa.eu/health/sites/health/files/files/eudralex/vol-4/2015-10_annex15.pdf
- 5Fourman, G. L., and M. V. Mullen. “Determining Cleaning Validation Acceptance Limits for Pharmaceutical Manufacturing Operations.” Pharmaceutical Technology 17 (1993): 54–54.
- 6Herman, A. M., and C. N. Jeffress. Informational Booklet on Industrial Hygiene, US Department of Labor, Occupational Safety and Health Administration, OSHA 3143 1998 (Revised). https://www.osha.gov/publications/OSHA3143
- 7International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use. “ICH Harmonised Guideline M7(R1): Assessment and Control of DNA Reactive (Mutagenic) Impurities in Pharmaceuticals to Limit Potential Carcinogenic Risk.” Final version adopted on 3 April 2023. https://database.ich.org/sites/default/files/ICH_M7%28R2%29_Guideline_Step4_2023_0216_0.pdf
- 8Melendez, E. “FDA Perspective on Multi-Product Facilities, Risk-Based Approach to Containment of Highly Hazardous Compounds.” Presented at the ISPE Conference/Containment Technology Forum, Tampa, FL, US, 25 February 2010.
- 9Naumann, B. D. “Setting Surface Limits (TLV-SLs).” Presented at the Organisation for Economic Co-operation and Development Online Workshop: “Approaches for Establishing Occupational Exposure Limits (OELs),” 21 and 24 October 2022.
- 10Suzuki, O. “Design Criteria for Barrier Isolation Systems and Containment Evaluation in Pharmaceutical Facilities.” Presented at the ISPE Conference, Amsterdam, Netherlands, 5–6 December 2001.
- 11Suzuki, O. “Design Criteria and Evaluation for Pharmaceutical Containment Systems.” Presented at the ISPE Conference, Philadelphia, PA, US, 11–12 September 2002.
- 12Suzuki, O. “Design Criteria and Evaluation for Pharmaceutical Containment Systems.” Presented at the ISPE Conference, Berlin, Germany, 2–3 December 2002.
- 13Maeck, R., Boehringer Ingelheim, “Evaluating Best Practice Containment & Navigating the High Potent Regulatory Landscape from EHS to GMP.” Presented at HPAPI Summit, Boston, MA, USA, 20–21 June 2018.
- 14Suzuki, O., et al. “Design Criteria and Evaluation for Pharmaceutical Containment Systems: Evaluation for Open Isolation Systems.” Pharmaceutical Engineering 23, no. 2 (March/April 2003): 66–78. https://ispe.org/sites/default/files/attachments/public/March-April-2003_1.pdf
- 15Suzuki, O., M. Takeda, K. Tanaka, and M. Numata. “Design Criteria and Evaluation for Pharmaceutical Containment Systems: Evaluation for Closed Isolation Systems.” Pharmaceutical Engineering 24, no. 5 (September/October 2004). https://ispe.org/sites/default/files/attachments/public/Sept-Oct-2004_0.pdf
- 16O’Connell, T., M. Coggins, V. Hogan, and M. Byrne. “Safety Evaluation of a Flexible Engineering Control System.” Pharmaceutical Engineering 26, no. 2 (March/April 2006). https://ispe.org/sites/default/files/attachments/public/March-April-2006.pdf
- 17Atmadi, A., Y. Nestiti, and Esco Micro Pte. Ltd. “Active Pharmaceutical Ingredients (API) Surrogate Containment Testing on ESCO POWDERMAX Powder Weighing Balance Enclosure.” Esco Lifesciences. https://escolifesciences.us/products/download/PW1_surrogate.pdf
- 18Liberman, D., C. Lockwood, M. McConnell-Meachan, E. J. McNally, H. Rahe, Kevin Shepard, et al. “Barrier Isolation Technology: A Safe and Effective Solution for Providing Pharmaceutical Development Facilities.” Pharmaceutical Engineering 21, no. 4 (July/August 2001): 20–8. https://ispe.org/sites/default/files/attachments/public/Jul-Aug-2001.pdf
- 19Hermann, F. “Production with Safe Handling of Highly Active Compounds (OEL 40ng/m3).” Presented at the ISPE 2010 Brussels Conference on Risk-Based Control Strategies in Pharmaceutical Industries, Brussels, Belgium, 22 September 2010.


