Holistic Control Strategies for Continuous Manufacturing

Innovative technologies such as continuous manufacturing (CM) bring speed, efficiency, and agility to pharmaceutical manufacturing together with enhanced process robustness and assurance of product quality. During CM, material is simultaneously charged and discharged into process unit operations. Similar to batch manufacturing, CM requires a comprehensive and holistic control strategy throughout the product life cycle to ensure, in a reproducible and consistent manner, the intended product quality at the time of release and throughout the product’s shelf life. A control strategy developed using a science and risk-based approach will ensure that reproducible product quality is achieved throughout the product life cycle, while maintaining the efficiency, robustness, and flexibility that CM has to offer.
Control strategy is described in ICH Q10 as “a planned set of controls, derived from current product and process understanding that assures process performance and product quality. The controls can include parameters and attributes related to drug substance and drug product materials and components, facility and equipment operating conditions, in-process controls, finished product specifications, and the associated methods and frequency of monitoring and control” 1 Fundamentally, control strategy expectations for CM are the same as those for batch manufacturing, namely that the manufacturing process is capable of consistently producing quality product. However, there is at least one notable difference between traditional batch manufacturing and CM. Many traditional batch manufacturing processes have unit operations that are designed to be well mixed (e.g., fermenters, bin blenders) and quality concerns are therefore related to variability with location or space. In contrast, a major quality concern for CM operations is related to temporal variability. Consequently, CM control strategies frequently incorporate process analytical technology (PAT) that provides information related to the product and/or process in real time.
CM provides an opportunity to build quality into the process design and control strategy, consistent with quality by design (QbD) principles.2 Because information is rapidly collected from CM systems, design space data can be readily obtained by varying settings and ranges of critical input parameters and analyzed by utilizing multivariate statistical design approaches (e.g., design of experiments [DoE]). A design space representing the multidimensional combinations and interactions of the critical attributes and parameters to demonstrate assurance of product quality is a potential element of a control strategy.3 Inline testing and modeling approaches, such as PAT, are other potential elements of a CM control strategy; these approaches provide an opportunity for real-time monitoring and control as well as real-time release testing (RTRT).
The overall control strategy for a CM process should be viewed in a holistic manner, with all elements of the control strategy working together to ensure product quality. There is no universal approach to a control strategy for CM, and multiple approaches can equally achieve quality and manufacturing goals. For example, a process that heavily incorporates in-process measurements and controls may have very little end-product testing, whereas an equally performing process may include fewer in-process measurements and extensive off-line analysis of finished product and process intermediates. Each manufacturer needs to decide on a control strategy approach that provides suitable mitigation of process risks while meeting business goals.
Simply having a well-developed control strategy alone is not sufficient to ensure manufacturing consistency and product quality. An effective pharmaceutical quality system (PQS) is integrated throughout the product life cycle to support the CM process.4 The process control strategy is continuously reassessed and enhanced, if necessary, following quality risk management (QRM) principles. Adjustments to the controls are made, if required, through continuous improvement identified from product and process performance monitoring and tracking. Product-release considerations, such as deviation management, diversion process, and alternate testing approaches, also are part of the PQS.
This article discusses the main elements associated with a control strategy for CM of solid oral dosage forms such as tablets and capsules. Although many of the control strategy considerations are the same as for a traditional batch process, CM control strategies emphasize the importance of raw material characterization and management, in-process testing (including PAT), and nontraditional batch-release approaches such as RTRT. Whereas the focus of this article is the processing of pharmaceutical solids, many of the control strategy aspects discussed here, such as system dynamics, are equally applicable to fluid-containing systems such as chemical reactions, cell culture processes, and separation or purification processes.
Raw Materials
Knowledge of the relationship between raw material attributes and the impact they have on product quality attributes is an important facet of product development, whether it is for CM or batch manufacturing processes. For a CM process, essential critical material attributes may differ from those for batch processing, with CM placing a stronger emphasis on dynamic powder characteristics, such as flowability, cohesiveness, and aeration. CM systems for solid oral dosage forms are heavily dependent on the ability of raw materials and intermediates to flow through the system. Therefore, the relationships of critical material attributes and process parameters to the critical quality attributes they affect should be understood. Understanding how critical raw material attributes impact ow, whether it involves drug substance or excipients being dispensed out of feeders or the dynamics of material movement through the equipment train, is of paramount importance.5 , 6
Physical properties of the drug substance and excipients, such as particle size, shape, and density, can affect feeder performance for raw materials. Poor feeder performance can have a negative impact on quality attributes such as assay or content uniformity, which can result in more material being diverted to waste and lower yields. When selecting a specific grade of an excipient, formulators should consider how different grades may affect manufacturability concerns such as compressibility and ow. If a batch process is to be converted to CM, the formulation ingredients used in the batch process should be reassessed to ensure that the material attribute specifications they possess will perform well in continuous processing.
The ability of the API to flow well is also essential to the smooth operation of CM for solid oral products. Particle engineering can be effective in crafting the physical properties of the API, if necessary, to provide appropriate ow characteristics. Where such efforts are not fruitful, alternative strategies can sometimes provide flowable material. In some cases, batch preblending of the API with a glidant excipient can provide material that flows well. In others, a drug product intermediate can be produced by spray-drying the API with excipients.
Raw material properties should be understood to ensure that the process is sufficiently robust to tolerate the introduction of different lots of raw materials, with no significant changes in the quality of the product or process performance. Changes in the particle size, shape, or density can impair ow and mixing, which leads to issues as the material traverses through the continuous processing equipment. This is of particular concern for extended campaigns that may consume different lots of the raw material over the course of the batch. A new batch of a raw material should not significantly affect the performance of material feeders because that could subsequently disrupt ow and alter the quality attributes of the drug product.
With appropriate understanding and controls, process operating conditions can be adjusted to compensate for the variability in a material attribute. When process controls are not successful in compensating for variation, material specifications should be adjusted to meet process needs. For example, to deliver a quality product, manufacturers may need tighter purchasing specifications for critical material attributes that are more restrictive than compendia requirements. In-process controls and specifications are both part of the control strategy.
Lot-to-lot variability of materials should be assessed, including the amount of variability that critical material attributes can tolerate without compromising product quality or manufacturability. Latent variable analysis on data from experimental results or quantitative values taken from the material certificates of analysis is one way to assess the amount of lot-to-lot variability in excipients.7 Multivariate monitoring of raw material properties within the PQS can determine when new variability is outside the ranges previously examined. Materials that are out of the range of previous experience may merit additional characterization and flow performance studies prior to being introduced into the manufacturing process.
Detecting and Controlling Disturbances
For many continuous operations, such as solids blending prior to tableting, disturbances in time can propagate through the system and affect a small portion of the manufactured material, potentially leading to production of out-of-specification material. It is essential that the control strategy be designed such that those disturbances do not occur, are not significant, or are detected and managed. A common method for managing disturbances in continuous systems is to isolate the material downstream from the point of the disturbance. 8 Understanding the system dynamics of a continuous system is essential for appropriately isolating such process disturbances and ensuring the “to be released” product is of the appropriate quality.
The goal for any manufacturing process, regardless of the technology or control strategy, is to operate within a state of control. The ICH Q10 defines state of control as “a condition in which the set of controls consistently provides assurance of continued process performance and product quality”. 1 Although this definition was not derived specifically for CM, it is fully applicable.
- 1 a b International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. “ICH Harmonized Tripartite Guideline: Pharmaceutical Quality System Q10.” Published June4, 2008. https://www.ich.org/fi leadmin/Public_Web_Site/ ICH_Products/Guidelines/Quality/Q10/Step4/Q10_Guideline.pdf
- 2International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. “ICH Harmonized Tripartite Guideline: Pharmaceutical Development Q8 (R2).” Published August 2009. https://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q8_R1/Step4/Q8_R2_Guideline.pdf
- 3International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. “ICH Endorsed Guide for ICH Q8/Q9/Q10 Implementation: ICH Quality Implementation Working Group Points to Consider (R2).” Published December6, 2011. http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/ Q8_9_10_QAs/PtC/Quality_IWG_PtCR2_6dec2011.pdf
- 4International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. “ICH Harmonized Tripartite Guideline: Quality Risk Management Q9.” Published November 9, 2005. https://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q9/Step4/Q9_Guideline.pdf
- 5Allison, G., Y. Cain, C. Cooney, T. Garcia, T. G. Bizjak, O. Holte, N. Jagota, et al. “Regulatory and Quality Considerations for Continuous Manufacturing, May20–21, 2014 Continuous Symposium.” Journal of Pharmaceutical Sciences 104, no. 3 (March 2015): 803–12. doi:10.1002/jps.24324
- 6Nasr, M. M., M. Krumme, Y. Natsuda, B. Trout, C. Badman, S. Mascia, C. Cooney, K. D. Jensen, A. Florence, C. Johnston, K. Konstantinov, and S. L. Lee.” Regulatory Perspectives on Continuous Pharmaceutical Manufacturing: Moving from Theory to Practice: September26–27, 2016, International Symposium on the Continuous Manufacturing of Pharmaceuticals.” Journal of Pharmaceutical Sciences 106, no. 11 (November 2017): 3199–206. doi:10.1016/j.xphs.2017.06.015
- 7Kushner, Joseph. “Utilizing Quantitative Certificate of Analysis Data to Assess the Amount of Excipient Lot-To-Lot Variability Sampled During Drug Product Development.” Pharmaceutical Development and Technology 18, no. 2 (March–April 2013): 333–42. doi:10.3109/10837450.2011.604784
- 8Lee, S. L., T. E. O’Connor, X. Yang, C. N. Cruz, S. Chatterjee, R. D. Madurawe, C. M. V. Moore, and L. X. Yu. “Modernizing Pharmaceutical Manufacturing: From Batch to Continuous Production.” Journal of Pharmaceutical Innovation 10, no. 3 (September 2015): 191–9.doi:10.1007/s12247-015-9215-8
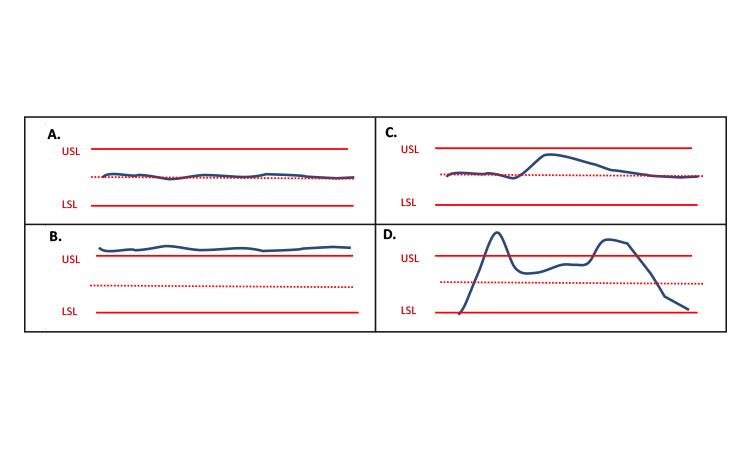
“Steady state” is a commonly used term related to CM and should not be confused with “state of control.” Generally, a steadystate condition can be described as a process state in which the quality attributes are kept approximately constant, or the rate of change with respect to time of those variables is approximately equal to zero over a relevant time span. Achieving steady state is neither su-fficient nor specifically necessary to be in a state of control. For example, a small disturbance in the process could cause the system to move out of a steady state while remaining in a state of control. Additionally, it is possible to have a system that is not changing (i.e., in steady state) and to have material that is not within specified ranges of quality attributes and thus not in a state of control. Figure 1 depicts different scenarios of steady state and state of control. Figure 1A shows a process that is at steady state and in a state of control; 1B shows a process that is at steady state and not in a state of control; 1C shows a process that is not at steady state but in a state of control; and 1D shows a process that is not at steady state and not in a state of control. Knowledge and process controls ensure that small disturbances will result in a controlled process that is making acceptable product, as shown in Figure 1C. Figure 1D illustrates when a large portion of the material is within specifications, but collecting it as good product would be inappropriate.
Residence Time Distribution Studies
A control strategy for CM should consider system dynamics aspects to provide consistent and reproducible product quality. For continuous systems, it is typical to measure or model a system’s residence time distribution (RTD), which describes the probability distribution of exit times for material entering the system. 9 The system’s RTD provides an indication of the system’s mixing efficiency, with the width of the distribution being proportional to the intensity of back-mixing in the system. The use of RTDs as a tool for traceability and process control is a regulatory expectation in many circumstances.10 By measuring and modeling the RTD, material can be tracked forward through the system as a function of time. This approach enables appropriate diversion of potentially nonconforming product resulting from instances where the system was outside of a state of control or beyond the control limits for any unit operation within the system. In addition, it also allows for traceability between incoming lots of raw actives and excipients and the final collected product.
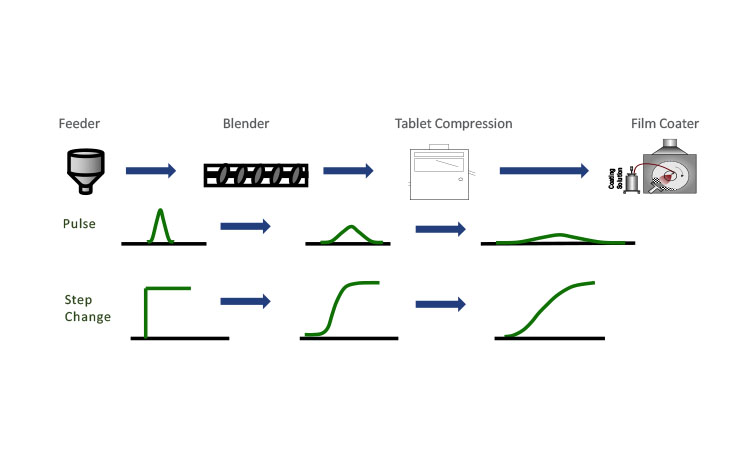
Conceptually, RTDs are somewhat straightforward, but the measurement of an RTD must be carefully considered if it is to be representative of the system’s dynamic residence time within the set operational limits.11 Two ways in which RTDs are commonly measured are with a pulse of a tracer or with a step change in composition (see Figure 2). In either case, it is crucial that the RTD determination conditions closely match the operational conditions. For tracer selection, a material with different physical properties may result in differences in how the tracer material flows relative to the operational blend, or it may change the blend properties overall. Similarly, for the step-change approach, where the concentration of API is monitored as the set point is changed, the stepped conditions will not be relevant if the resulting blend properties are significantly different. RTD characterization and validation efforts should evaluate both the sensitivity of the RTD to system variation and the validity of the RTD experimental and modeling assumptions. Instances where one set of blend properties vary significantly enough from another could lead to erroneous results for the RTD parameters.
- 9Gao Y., A. Vanarase, F. Muzzio, and M. Ierapertritou. “Characterizing Continuous Powder Mixing Using Residence Time Distribution.” Chemical Engineering Science 66, no. 3 (February 2011): 417–22. doi:10.1016/j.ces.2010.10.045
- 10Kourti T., R. Madurawe, K. Brorson, D. Doleski, D. Hernán Pérez de la Ossa, J. Hu-Primmer, C. Airiau, et al. “ISPE 2016 Continuous Manufacturing Conference Highlights.” Pharmaceutical Engineering 37, no. 3 (May/June 2017): 35–42.
- 11Escotet-Espinoza M., S. Moghtadernejad, S. Oka, Z. Wang, Y. Wang, E. Román A, Schäfer, P. Cappuyns, et al. “Effect of Material Properties on the Residence Time Distribution (RTD) Characterization of Powder Blending Unit Operations. Part II of II: Application of Models.” Powder Technology 344 (February 15, 2019): 525–44. doi:10.1016/j.powtec.2018.12.051
Models utilizing the RTD can determine which disturbances will dampen out and not affect product quality and which disturbances dictate that material be diverted from the system. The models can further inform how much material needs to be isolated. Sampling frequency and measurement capability are important considerations in these calculations to ensure appropriate detection of disturbances.
Refill Schedule
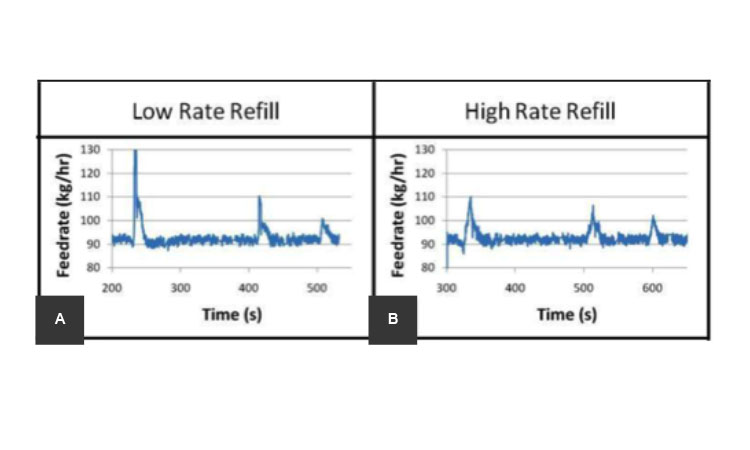
Refilling of the feeders, which introduce material into the process, is an inherent part of CM processes of solids. In nearly all cases, feeder refills introduce process disturbances of varying degrees.12 Refill schedules can be optimized to minimize their effect on the system’s process dynamics. Tools such as RTDs can be used to determine the conditions in which a refill may put product quality at risk, and how much (or how long) at-risk product may need to be segregated from the final collection stream. A refill schedule should be determined for adequate performance of the feeders. Figure 3 shows an example of disturbances for low and high feeder refill rates; the lower refill rate (Figure 3A) introduces a higher quantity of material, thus causing a greater disturbance than a more frequent higher refill rate (Figure 3B).
Start-Up, Shutdown, and Pauses
Start-up and shutdown of continuous systems are process conditions where the system is not intended to be operating at a steady state and process conditions are known to be changing. This does not imply that the system is not in a state of control. If the process dynamics of start-up and shutdown have been appropriately characterized, a state of control can be maintained throughout the manufacturing process from start-up through shutdown. The value of the effort to demonstrate a state of control during the short periods of start-up and shutdown depends on product value and throughput. For large-volume commodity products operating at high throughputs and for long periods of time, lost material from start-up or shutdown may be insignificant.
Material Diversion
Control strategies for CM are often designed to divert potentially nonconforming material when the process is not in a state of control. Material diversion can be planned or unplanned. An example of planned diversion is the removal of any nonconforming material that is generated during start-up, shutdown, or process pauses. Unplanned diversion or process interruption may be necessary when a trend or change in the material’s characteristics or the process performance is detected. The PQS should include written and approved procedures that state how the process is to be shut down or paused, explain the circumstances in which an investigation into the root cause of the problem is required, outline when/how production can resume, and provide the steps for restarting the process.
The ability to perform diversions and partial lot rejections for CM is somewhat unique in pharmaceutical operations. In batch manufacturing, a lack of a state of control for the process commonly results in nonsegregable, nonconforming material and subsequent total batch failure. However, because of the high level of process understanding, traceability, and control in CM, portions of the batch containing potentially nonconforming material can be readily segregated from the remainder of the produced material that is verified to be of known acceptable quality. In most cases, the segregated portion of the batch will be rejected and discarded. However, in some cases, an investigation may reveal false signals leading to the diversion decision, such as data from a fouled measurement probe. In such a case, it could be justifiable to reintroduce the diverted material back into the acceptable product. Clear instructions for these types of decisions, as well as guidance on what supporting data are required, should be captured in the PQS.
A high or atypical number of diversion events or a large amount of rejected material can reduce confidence in the quality of the acceptable portion of the batch. Failure to comply with defined minimum yields for the batch may prohibit its release for commercial sale.
- 12Nowak, S. “Improving Feeder Performance in Continuous Pharmaceutical Operations.” Pharmaceutical Technology 40, no. 10 (October 2, 2016): 68–73. http://www.pharmtech.com/improving-feeder-performance-continuous-pharmaceutical-operations
Process Measurements and Controls for CM
In general, process measurements and controls for CM can be described as a combination of in-process controls, process performance monitoring, equipment controls, and facility controls. A combination of all of these ensures that the manufacturing process remains in a state of control and consistently produces quality product. In-process controls are the checks during production that appropriately adjust the process to ensure conformance to specifications;13 these are also known as “in-process tests”.14 Process performance monitoring is not specifically defined in ICH guidelines, but it generally can be understood to be measurements that are used to assess process performance or consistency; these measurements can provide signals related to future quality issues. Equipment controls oversee the operation of specific types of equipment; examples include automatic adjustments or shutoffs. Typically, equipment controls are independent of the mode of manufacture. Facility controls for CM, such as room temperature and relative humidity, are the same as for traditional batch manufacturing.
The ICH Q10 definition for control strategy is very broad and includes many control strategy elements that are not included in the regulatory dossier. For example, in-process controls are typically discussed as part of the registered control strategy, whereas process performance monitoring, equipment controls, and facility controls are not included in the dossier. Aspects such as equipment operating conditions and frequency of monitoring are elements of the control strategy but are not typically included in the dossier.
PAT for Monitoring and Control
PAT can be incorporated into CM control strategies in many ways, such as by measuring product attributes in the process or by determining process performance. Measurement of product attributes can be direct or inferential and can be used for actionable control decisions or for monitoring purposes. Measurements can be taken at frequencies relative to the level of risk for the attribute, such as during changeover to a new lot of a drug substance or excipient.
A wide variety of PAT tools and approaches can be used to directly measure product attributes. The most common PAT tools for measuring product attributes in solids are spectroscopic, including near-infrared spectroscopy (NIR) and Raman spectroscopy. Although both spectroscopy tools can provide concentration measurements of multiple species, Raman spectroscopy can additionally provide information on solid-state characteristics, such as polymorphism.15 Regardless of the PAT tools used, the purpose is the same: to provide real-time measurements of the system such that timely decisions can be made.
Process data, such as process parameter values, material attributes, and data from sensors, can be analyzed in a multivariate manner to determine process performance and process consistency. Multivariate approaches such as multivariate statistical process control can often reveal discrepant performance that univariate trending would not identify and can aid in early diagnosis of process or equipment failures.16
Multivariate analysis also can be applied as a parametric approach to infer product quality data indirectly from process information. In a parametric, or “soft sensor” approach, a broad array of data from the process and materials is correlated in a multivariate fashion to help predict product quality attributes. Parametric PAT approaches are evolving from both a technical and regulatory perspective.
Many PAT systems, such as spectroscopy, require life-cycle maintenance and updates to the underlying models to ensure that they continue to function as intended.17 All models used in-process control and monitoring are expected to be managed and maintained within the PQS. In some regions, regulatory reporting may be required for updates to models related to measurement of product quality. Typically, models for process monitoring of performance or consistency are maintained within the PQS without regulatory reporting.
PAT can be incorporated into CM control strategies in many ways, such as by measuring product attributes in the process or by determining process performance.
Process Control Approaches
A robust control strategy for CM will emphasize controlling the product quality in response to potential variations in the process and equipment conditions over time, properties of incoming raw materials, or external environmental factors. The control strategy elements support the continued state of control, proper product collection, and product quality. The dynamic and integrated nature of CM increases the benet of enhanced control strategies that employ control elements other than the traditional offline end-product testing.
According to Lee and colleagues,8 control strategy implementation can be categorized into three levels based on the robustness, flexibility, and complexity of control elements and will depend on many factors, including the desired product performance, manufacturing process, and process dynamic characteristics (e.g., product heterogeneity and mixing patterns). The base level (Level 3), which is commonly used in traditional batch manufacturing, typically relies on tightly constrained material attributes and process parameters with extensive end-product testing to ensure product quality. The intermediate level (Level 2) has more flexibility in raw material attributes and process parameters through utilization of an established design space. The ultimate level (Level 1) has active process controls to monitor the quality attributes and adjust the process in real time. In practice, a control strategy for CM may display a combination of control elements at any of the three levels, provided that the risks to product quality are effectively controlled and mitigated.
Through flexible operations, where extensive process information is obtained during manufacturing, a Level 1 process can adjust for variability in raw material and equipment conditions to ensure on-target product is produced. In contrast, traditional process control schemes (Level 3) have predetermined, fixed operating points or ranges. In these cases, the quality of the product is only determined after the process is complete, with little or no opportunity to make corrections or adjustments.
CM control systems often use feedback and/or feedforward controls to adjust process parameters. An example of feedforward control is use of a loss-in-weight feeder data with an RTD model to predict the concentration downstream and determine diversion times for nonconforming material. An example of feedback control is adjustment of feeder ow rates based on inline blend NIR data. Other control configurations such as cascade control and ratio control are also possible.
No single set of controls or control strategies is appropriate for every continuous pharmaceutical manufacturing operation. The process controls should be based on the specific product, formulation, and process design as well as the associated risks to product quality. Processes with poorly owing material or environmental sensitivity may need more in-process measurements or controls than is required for products and processes with fewer failure modes.
Alternative control strategies are also allowable3 and, in some cases, expected. For example, in the event of a failure of a PAT control, a contingency plan should be developed to allow manufacture of the batch to be completed.18 The contingency plan should be capable of detecting perturbations and, as appropriate, enable diversion of material that is out of specification; the diversion may need to be performed manually. Appropriate statistically based sampling plans and acceptance criteria are needed to provide confidence that the quality of the batch is acceptable for release. Drug product and intermediates may be tested with either offline PAT or traditional analytical chemistry approaches.
Scale-Up of CM
In traditional batch manufacturing, the term “scale-up” usually refers to the manufacture of greater quantities of materials through use of physically larger equipment. For CM, however, manufacturing of greater quantities of material usually occurs simply by running the same equipment for a longer time and/or at a faster rate.
CM rigs are often referred to in terms of throughput rather than equipment size or scale. Throughput is the amount of material processed by a system and is typically represented in units of mass per time, such as kg/hr. In some cases, manufacturing lines will be named based on the range of achievable throughputs (i.e., a 25 kg/hr or 50 kg/hr line), but, in reality, the throughput on a given line will likely be formulation dependent. On a specific system, the upper and lower bounds of throughput or achievable ow rate while a state of control is maintained may be different for each formulation or material attribute characteristic. Therefore, process development and validation activities are typically performed at a specific throughput or set of throughputs.
The control strategy elements for CM primarily remain the same for different batch sizes or “scales.” However, there are some time-dependent aspects of longer run times that should be considered in the development of the control strategy and validation plan; these include, but are not limited to, effects of equipment wear, potential for accumulation of material in the system, and possibility of microbial growth.
Product Specifications and Release Considerations
Product specifications are part of the control strategy and address the quality attributes’ ranges and targets that must be achieved to ensure the safety and efficacy of the product. Although the quality standards for the release of intermediates and finished products remain the same as those applied to batch processes, nontraditional analytical methods and acceptance criteria can be used to demonstrate
compliance with quality attributes. In a traditional approach (Level 3 control), end-product testing to product specification provides the primary assurance of product quality. In a more advanced control strategy, the primary assurance of quality is through process controls, including in-process tests, and the primary purpose of any end-product testing is to confirm that the process is operating as intended.
Different approaches can be used for the release of intermediates and finished product, including RTRT, traditional methods, or a hybrid approach. Continuous processes typically employ more PAT methods than batch processes, which facilitate the use of RTRT for in-process controls and release testing. Although advanced control strategies are frequently used in CM, the use of traditional approaches (i.e., off-line testing of in-process samples and end-product testing) for batch release remains a viable option, especially for backup systems in the event of PAT failures.
Real-Time Release Testing
RTRT is used to evaluate and ensure the quality of in-process materials and/or final product based on process data that typically include a valid combination of measured raw material attributes and process controls.2 In this context, implementation of RTRT to CM processes requires establishing clear relationships between the final product’s critical quality attributes and the control elements incorporated into the process (e.g., quality attributes of raw and in-process materials, process parameters).
NIR spectroscopy is commonly used in CM of solid oral dosage forms and lends itself readily to an RTRT approach for identity, assay, and content uniformity (e.g., through NIR analysis of the blend prior to compression). Content uniformity can be calculated from the combination of NIR blend potency and weight uniformity of the dosage forms. A more complex example of an RTRT approach is a dissolution model based on the real-time measurements of in-process material or end-product quality attributes (e.g., drug concentration, tablet hardness, weight, and particle size distribution) and use of an appropriate mathematical model to predict the tablet dissolution performance.19 When using an RTRT approach for product release, redundant testing for that particular attribute (i.e., wet chemistry) can be eliminated. However, traditional laboratory testing methods must be available to aid in post-manufacturing analysis.
An RTRT approach warrants careful consideration of the sampling strategy that accounts for equipment dynamics and the RTD for material passing through the system. The selected sample size and frequency should be representative of the batch and justified statistically to provide an adequate confidence level and coverage. Given the high frequency of data collection, appropriate statistical methods for large sample size can increase the confidence level that the batch conforms to the desired quality.20
In the event of PAT equipment failure, established alternative procedures can be used for process monitoring and batch release.18 These procedures could include end-product testing or the use of surrogate measurements to ensure that products demonstrate an acceptable level of quality.
Hybrid Release Specifications
As previously stated, an approach that combines traditional and RTRT release methods can also be used. For instance, identity, assay, and blend/content uniformity may be determined by NIR, with dissolution and impurities being determined through traditional laboratory analysis. Some tests, such as microbial content, may not be possible for PAT, which may prevent a full RTRT approach to release testing.
Traditional Release Specifications
Traditional product-release tests can be used with CM and, in some instances, may be preferred. For example, if the manufacturing process is a hybrid of both batch and CM processes, or if the final step (e.g., film coating) is conducted as a batch process, it may be easier to perform product-release testing on manual samples using traditional analytical methods and acceptance criteria. For any control strategy approach, sufficient assurance of quality should be justified through appropriate raw material specification, process parameter controls, in-process tests, and end-product testing.
Conclusions
Control strategies for CM should be holistic in their design, appropriately utilizing a combination of raw material specifications, in-process tests, process monitoring and controls, and end-product testing to specifications. There is no one-size-fits-all approach to CM control strategies. Each process and product has its own risks that the control strategy mitigates through science- and risk-based approaches along with a robust PQS. Although advanced controls are commonly used in CM for solid oral dosage forms, they may not necessarily be required, depending on the level of process understanding, manufacturing experience, and specific process risks. A comprehensive and holistic view of all elements of the control strategy provides continued assurance of product quality over the product and process life cycle.
- 13International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. “ICH Harmonized Tripartite Guideline: Good Manufacturing Practice Guide for Active Pharmaceutical Ingredients Q7.” Published November10, 2000.https://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q7/Step4/Q7_Guideline.pdf
- 14International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. “ICH Harmonized Tripartite Guideline: Development and Manufacture of Drug Substances (Chemical Entities and Biotechnological/Biological Entities) Q11.” Published May 1, 2012. https://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q11/Q11_Step_4.pdf
- 15Romero-Torres, S., J. Huang, and P. Hernandez-Abad. “Practical Considerations on PAT Analyzer Selection—Raman vs. NIR Spectroscopy.” American Pharmaceutical Review no. 12 (December 1, 2009): 5–10.
- 16Kourti, T. “Process Analytical Technology Beyond Real-Time Analyzers: The Role of Multivariate Analysis.” Critical Reviews in Analytical Chemistry 36, no. 3–4 (2006): 257–78.doi:10.1080/10408340600969957
- 17Wise, B., and N. Gallagher. “The Process Chemometrics Approach to Process Monitoring and Fault Detection.” Journal of Process Control 6, no. 6 (December 1996): 329–48.doi:10.1016/0959-1524(96)00009-1
- 8
- 3
- 18 a b European Medicines Agency. “Guideline on Real Time Release Testing (formerly Guideline on Parametric Release).” Published March 29, 2012. https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-real-time-release-testing-formerly-guideline-parametric-release_en.pdf
- 2
- 19Pawar, P., Y. Wang , G. Keyvan, G. Callegari, A. Cuitino, and F. Muzzio. “Enabling Real Time Release Testing by NIR Prediction of Dissolution of Tablets Made by Continuous Direct Compression (CDC).” International Journal of Pharmaceutics 512, no. 1 (October15, 2016):96–107. doi:10.1016/j.ijpharm.2016.08.033
- 20European Directorate for the Quality of Medicines. “Chapter 2.9.47: Demonstration of Uniformity of Dosage Units Using Large Sample Sizes.” In European Pharmacopeia, 9th ed.2019. http://online6.edqm.eu/ep908


